Fenylketonuri - klassiska tecken på ärftlig överföring och dietterapi
Innehåll
- 1Hur manifesteras fenylketonuria
- 2Mekanism för sjukdomsutveckling
- 3Fenylketonuria hos barn
- 4Symtom på sjukdomen
- 5Orsaker och triggers
- 6Diagnos
- 7Behandling av klassisk fenylketonuri
- 8Funktioner för näring hos nyfödda ochdietterapi
- 9Kost för förskolebarn och skolbarn
- 10Produktgrupper med PKU
- 11Hur man kontrollerar nivån av fenylalanin i blodet
- )12Video
Sjukdomar vars förekomst är relaterad till defekter i den genetiska cellulära apparaten - fenylketonuria - ingår i en liten lista över ärftliga sjukdomar som kan behandlas.Pionjären för denna sjukdom var en norsk läkare IA Felling, det upptäcktes senare att en enda gen som kallas fenylalaninhydroxylasgenen (den långa armen i den 12: e kromosomen innehållande upp till 4,5% av allt cellulärt DNA-material) var ansvarig för utvecklingen och förloppet av sjukdomen.Ärftlig defekt leder till partiell eller fullständig deaktivering av leverenzymet fenylalanin-4-hydroxylas.
Hur fenylketonuri sjukdom upptäcks
Ärvt fenylketonuri (PKU) sjukdom leder till kronisk förgiftning av kroppen med giftiga ämnen som bildas som ett resultat av försämrad aminosyrametabolism och processhydroxylering av fenylalanin.Permanent berusning orsakar skador på centrala nervsystemet (CNS), varvid en manifestation är en progressiv minskning av intelligensen (fenylpyruvic oligofreni).
Fells sjukdom manifesteras i den överdrivna ansamlingen i kroppen av fenylalanin och produkterna från dess felaktiga ämnesomsättning.Andra faktorer för utveckling av fenylketonuri inkluderar nedsatt aminosyratransport över blod-hjärnbarriären, låga neurotransmitterantal (serotonin, histamin, dopamin).I avsaknad av en snabb behandling av sjukdomen leder till mental retardering och kan orsaka barnets död.

Mekanismen för sjukdomsutveckling
Orsaken till genstörningar är ett metaboliskt block som förhindrar bildandet av fenylalanin-4-hydroxylas (ett enzym,vilket är ansvarigt för omvandlingen av fenylalaninaminosyran till tyrosin).Proteinogen aminosyratyrosin är en integrerad del av proteiner och melaninpigment, så det är ett nödvändigt element för att fungera i alla kroppssystem, och dess brist leder till fermentopati.
Inhiberingen av metabolitbildning orsakad av mutationsinaktivering av enzymet är aktivering av hjälpmetaboliska vägar för fenylalanin.Aromatisk alfa-aminosyra, som ett resultat av defekta metaboliska processer, delar sig in i toxiska derivat, som under normala förhållanden inte bildar:
- fenylpyruvinsyra (fenylpyruvat) - en fet-aromatisk alfa-ketosyra, dess bildningleder till myelinisering av neuronala processer och demens;
- fenylmelkesyra är en produkt som bildas under återvinning av fenylpyruvinsyra;
- fenyletylamin - den initiala föreningen för biologiskt aktiva sändare av elektrokemiska impulser, ökar koncentrationen av dopamin, adrenalin och norepinefrin;
- Ortofenylacetat är en giftig substans som orsakar metaboliska störningar i hjärnan.
Medicinsk statistik indikerar att en patologiskt förändrad gen finns i 2% av befolkningen, men den visar sig inte på något sätt.Den genetiska defekten överförs till barnet från föräldrarna endast i närvaro av sjukdomen hos båda parterna, där barnet i 50% av fallen blir bäraren av den muterade genen och förblir frisk.Sannolikheten för att fenylketonuri hos nyfödda leder till sjukdom är 25%.
Efter vilken typ ärvs
Fells sjukdom är en genetisk avvikelse som ärvs från en autosomal recessiv typ.Denna typ av arv innebär att utvecklingen av tecken på en medfödd sjukdom endast kommer att ske när en defekt genkopia från båda föräldrarna som är heterozygota bärare av den förändrade genen ärvs från barnet.
Utvecklingen av en medfödd sjukdom i 99% av fallen orsakas av en mutation av genen som är ansvarig för kodning av enzymet som ger syntes av fenylalanin-4-hydroxylas (klassisk fenylketonuri).Upp till 1% av genetiska sjukdomar är relaterade till mutationsförändringar som inträffar i andra gener som orsakarbrist på dihydropteridinreduktas (PKU typ II) eller tetrahydrobiopterin (PKU typ III).
Fenylketonuria hos barn
Den klassiska formen av genetisk sjukdom hos barn manifesterar sig i de flesta fall utåt, med början vid åldern 3-9 månader.Nyfödda med en defekt gen, ser friska ut, en särdrag är babyens specifika vana (utseende).Den uttryckta symptomatologin förekommer inom 6-12 månader efter födseln.
Typ II PKU kännetecknas av det faktum att de första kliniska symtomen uppträder 1,5 år efter födseln.Tecken på sjukdomen försvinner inte efter diagnosen genetiska avvikelser och inledningen av dietterapi.Denna typ av medfödd sjukdom leder ofta till ett dödligt resultat under 2-3 år av barnets liv.De vanligaste symptomen på typ II PKU är:
- allvarliga psykiska störningar;
- hyperreflexi;
- nedsatt motorisk funktion av alla extremiteter;
- syndrom av okontrollerade muskelkontraktioner.
Kliniska tecken på mutationsförändringar i typ III-gener liknar typ II-sjukdom.Tetrahydrobiopterinbrist kännetecknas av en triad av specifika symtom:
- en hög grad av mental retardering;
- skalens storlek minskas tydligt relativt andra kroppsdelar;
- muskelspasticitet (fullständig förlust av rörligheten i extremiteterna är möjlig).

Manifestationer av Fells sjukdom
I kliniska studier och observationer har det föreslagits atttoxiska derivat av fenylalaninmetabolism orsakar en minskning av den intellektuella förmågan, som är progressiv till sin natur och kan leda till demens (oligofreni, idioti).Bland de troliga orsakerna till irreversibla störningar i hjärnaktiviteten av de mest rimliga anses vara orsakade av en minskning av nivån av tyrosinbrist på neurotransmittorer som överför impulser mellan neuroner.
Det exakta förhållandet mellan orsak och verkan mellan ärftlig sjukdom och hjärtsjukdomar har ännu inte identifierats, liksom mekanismen för utveckling på grund av fenylketonuri i sådana mentala tillstånd som ekopraxi, echolalia, hårda attacker och irritabilitet.Resultaten av analysen indikerar att fenylalanin har en direkt toxisk effekt på hjärnan, vilket också kan orsaka en minskning av intelligensen.
Struktur och fenotypiska egenskaper
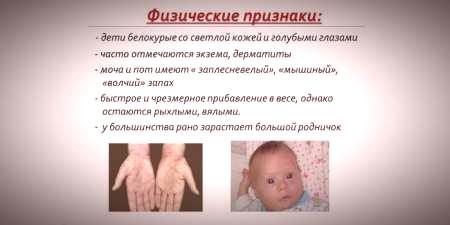
Med tanke på att hud- och hårpigmentmättnad beror på tyrosinnivån i hepatocyt-mitokondrier och fenylketonuri leder till stopp av fenylalaninomvandling, har patienter med denna sjukdom särskiljningsförmågarecessiva tecken).Ökad muskelton orsakar uppkomsten av avvikelser i kroppens struktur - det blir dysplastiskt.Utmärkande yttre egenskaper hos fenylketonuri inkluderar:
- hypopigmentering - ljus hud, ljusblå ögon, missfärgat hår;
- cyanos av extremiteter;
- minskad huvudstorlek;
- kroppens specifika position - när barnet försöker stå eller sitta antar barnet en "skräddarsydd" ställning (armar och ben böjda vid lederna).
Symptomsjukdom
Med snabb upptäckt behandlas Fells sjukdom framgångsrikt genom att justera näring och barnets utveckling sker enligt hans åldersgrupp.Svårigheten att upptäcka en genmutation är att tidiga tecken är svåra att upptäcka även av en erfaren barnläkare.Svårighetsgraden av symtomen på medfödd sjukdom ökar när barnet blir äldre, eftersom konsumtionen av proteinmat bidrar till utvecklingen av CNS-störningar.
Tecken på nyfödda
Under de första dagarna av barnets liv är tecken på onormala avvikelser svåra att upptäcka - barnet uppför sig naturligt, det finns inga utvecklingsförseningar.Symtomen på sjukdomen börjar dyka upp för första gången 2-6 månader efter födseln.Föräldrarna bör vara uppmärksamma på barnets beteende, som kännetecknas av låg aktivitet, slöhet, eller tvärtom, ångest, hyper-excitability.
När amningen börjar börjar nyfödda med mjölk komma in i den nyfödda kroppen med mjölk, vilket är en katalysator för uppkomsten av de första tecknen, vilket tydligt indikerar att sjukdomen har börjat utvecklas.Specifika kliniska manifestationer av sjukdomen inkluderar:
- konstant kräkningar (ofta anses medfödd förträngning av målvakten);
- frekvent kräkningar;
- inget svar på yttre stimuli;
- muskeldystoni (minskad muskelspänning);
- konvulsivt syndrom (kramper av epileptisk eller icke-epileptisk natur).
Symtom hos barn efter 6 månader
Om manifestationen av genetisk sjukdom inte ärinträffade (eller observerades inte) under de första 6 månaderna efter barnets födelse, och efter denna period är det redan möjligt att exakt bestämma förseningen i psykomotorisk utveckling.Symtom på genetiska störningar orsakade av enzymbrist hos barn äldre än sex månader är:
- minskad aktivitet (upp till fullständig likgiltighet);
- brist på självbehörighet, sittplatser;
- en märklig "mus" -lukt i huden (lukten av mögel är resultatet av utsöndring av giftiga fenylalaninderivat genom svettkörtlar och urin);
- förlust av förmågan att visuellt känna igen föräldrarnas ansikten;
- skalning av huden;
- dermatit, eksem, sklerodermi.
Progression av sjukdomen i avsaknad av behandling i barndomen
Om utvecklingsavvik inte upptäcktes i barndomen och lämplig behandling inte utfördessjukdomen börjar utvecklas aktivt och leder ofta till funktionshinder.Brist på terapi i ett tidigt stadium av sjukdomen får följande symtom att utvecklas vid 1,5 års ålder:
- mikrocefali (minskad hjärnstorlek);
- prognathia (förskjutning av den övre tandvården framåt);
- sent tandläkare;
- emaljhypoplasi (tunnare eller fullständig frånvaro av tandemaljen);
- förseningen i språkutvecklingen fram till fullständigt frånvaro av tal.
- 3, 4 grad av oligofreni (mental retardering, mental retardering);
- medfödda hjärtfel (defekter i hjärtmuskelns struktur, delar av hjärtat,stora fartyg);
- störningar i det autonoma systemet (akrocyanos, överdriven svettning, arteriell hypotension);
- förstoppning.
Orsaker och provocerande faktorer
För manifestationen av en autosomal recessiv mutation måste en defekt gen ärvas från båda föräldrarna.Genetiska sjukdomar av denna typ förekommer med samma frekvens hos nyfödda pojkar och flickor.Patogenes av PKU orsakas av försämrad metabolism av fenylalanin, som kan förekomma i tre former.Dietterapi underkastas endast klassisk fenylketonuri typ I.
Atypiska former av sjukdomen kan botas genom att justera näring.Dessa avvikelser orsakas av brist på tetrahydropterin, dehydropterinreduktas (sällan - pyruvyltetrahydropterinsyntas, guanosin-5-trifosfat och annat).De flesta fall av dödliga fall registreras bland patienter med sällsynta variationer av PKU, med de kliniska manifestationerna av alla former av sjukdomen liknande.Risken för att få ett barn med en muterad fenylalaninhydroxylasgen ökar om föräldrarna är nära släktingar (i nära besläktade äktenskap).
Diagnos
Vid misstänkta genetiska störningar ställs diagnosen på grundval av en uppsättning data som erhållits från studier av medicinsk historia - släktuppgifter, resultaten från kliniska och medicinska genetiska studier.För snabb upptäckt av medfödda sjukdomar (PKU, cystisk fibros, galaktosemi etc.) utvecklades obligatoriskt massprogramlaboratorieundersökning av alla nyfödda (nyfödda screening).
Om förväntade föräldrar är medvetna om bäraren av den muterade genen erbjuder modern medicin sätt att upptäcka en defekt vid graviditetsstadiet (prenatal diagnos av fostret med invasiv metod).För uppdelning av fenylketonuri i typer efter svårighetsgrad används en villkorad klassificering baserad på nivån av fenylalanin i en fibrinogenfri blodplasmavätska:
- Tung fenylketonuri - 1200 μmol /l.
- Genomsnitt - 60-1200 umol /l.
- Ljus (ingen behandling krävs) - 480 μmol /L.
Screening Test
Upptäckten av genetiska avvikelser sker i flera steg.I det första steget på modersjukhuset provas alla spädbarn i 3-5 dagars liv perifert blod (från fem) för forskning.Materialet appliceras på ett pappersformulär och skickas till det biokemiska laboratoriet där det är biokemisk analys.I det andra steget av screeningstestet bestäms koncentrationen av normal fenylalaninkoncentration.

Om inga patologiska förändringar upptäcks är diagnosen klar och barnets kort registreras.Vid avvikelser från normen skickas resultaten av diagnosen till barnläkaren för att säkerställa en mer exakt undersökning av blodprovet hos den nyfödda.Barnets hälsa beror på att samtliga åtgärder för att upptäcka avvikelser genomförs i rätt tid och korrekt.Om diagnosen bekräftas efter upprepad screening kommer barnets föräldrar att göra detskickas till en klinik för barngenetik för behandling.
Analyser och studier för att bekräfta diagnos
Återdiagnos vid detektion under det primära screeningtestet av avvikelser utförs genom att skicka in testerna igen.Förutom att bestämma innehållet av fenylalanin i blodet till metoder för diagnos av PKU hos barn och vuxna inkluderar:
- Avverkningstest - bestämning av fenylpyruvinsyra i urinen genom att tillsätta järnklorid till biomaterialet (färgning i blågrön färg);
- Guthrie-test - bedömning av graden av mikroorganismernas respons på metabolismen eller enzymerna i patientens blod;
- kromatografi - studien av de kemiska egenskaperna hos ämnen fördelade mellan två faser;
- fluorimetri - bestrålning av biomaterial genom monokromatisk strålning för att bestämma koncentrationen av ämnen som finns däri;
- elektroencefalografi - diagnos av hjärnans elektriska aktivitet;
- Magnetisk resonansavbildning är excitering av atomkärnorna i cellerna genom elektromagnetiska vågor och mätningen av deras svar.
Behandling av klassisk fenylketonuri
Kärnan i fenylketonuri-terapi är en begränsning av konsumtionen av produkter som är källan till animaliska och vegetabiliska proteiner.Den enda metoden för framgångsrik behandling är dietterapi, vars tillräcklighet utvärderas med innehållet av fenylalanin i serumet.Maximal nivå av aminosyror hos patienter i olika åldersgrupperär:
- hos spädbarn och barn upp till 3 år - upp till 242 umol /l;
- i förskolebarn upp till 360 umol /l;
- hos patienter mellan 7 och 14 år - upp till 480 μmol /l;
- hos ungdomar - upp till 600 μmol /l.
Kostens effektivitet beror på vilket stadium av sjukdomen dieten korrigeras för.Vid tidig diagnos av medfödd patologi föreskrivs dietterapi från den åttonde veckan i livet (efter denna period påbörjas irreversibla förändringar redan).Frånvaro av snabba åtgärder leder till komplikationer och en minskning av intelligensnivån med 4 poäng under en månad från födseln till behandlingens början.

Med tanke på att den terapeutiska dieten för fenylketonuri ger fullständig uteslutning från kosten för animaliskt protein, finns det ett behov av att använda andra källor till essentiella aminosyror, såväl som vitaminer i grupp B, kalcium -och fosforinnehållande mineralföreningar.Produkterna som ska användas som proteinfria tillskott inkluderar:
- proteinhydrolysat (Amigen, Aminazol, Fibrinosol);
- innehåller inte fenylalaninblandningar mättade med essentiella aminosyror - Tetrafen, fenylfri.
Utöver botande åtgärder för att eliminera orsaken till nedsatt kroppsfunktion bör symptomatisk behandling genomföras för att eliminera talfel och normalisera koordinationen av rörelser.Komplex terapi inkluderar fysioterapiprocedurer, massage, hjälp av en logoped, en psykolog, utför gymnastiska övningar.I vissa fall i samband med dietterapivisar användningen av antikonvulsiva medel, nootropiska och vaskulära läkemedel.
Särdrag vid behandling av atypiska former
Fenylketonuri typ II och III kan inte behandlas med lågproteindiet - nivån av fenylalanin i blodet förblir oförändrad medan begränsningen av proteinflödet i kroppen eller kliniska symtom fortskrider även med minskningen av aminosyranivån.Effektiv terapi av dessa former av sjukdomen utförs med användning av:
- tetrahydrobiopterin - faktor för det drabbade enzymet;
- syntetiska analoger av tetrahydrobiopterin - dessa ämnen penetrerar bättre blod-hjärnbarriären;
- substitutionsbehandlingsläkemedel - eliminerar inte orsaken till fenylketonuri, men stöder kroppens normala funktion (Levodopa tillsammans med Carbidofa, 5-oxytryptofan, 5-formyltetrahydrofolat);
- hepatoprotectors - stödjer leverfunktion;
- antikonvulsiva medel;
- introduktion av fenylalaninhydroxylasgenen i levern är en experimentell metod.
Nyfödd närings- och dietterapi
Amning är tillåtet under det första leveåret för ett barn med PKU, men bör begränsas.Upp till 6 månader är det acceptabla intaget av fenylalanin 60-90 mg per 1 kg babyvikt (100 g mjölk innehåller 5,6 mg fenylalanin).Från tre månader bör barnets diet gradvis utvidgas och införa fruktjuicer och puréer i den.
Barn från 6 månaders ålder får lägga in dieten med grönsaker, gröt (sago), proteinfriKiselev.Efter 7 månader är det möjligt att ge baby-low-protein pasta, från 8 månader - bröd, som inte innehåller protein.Den ålder då proteinet i det sjuka barnets kropp ska begränsas har inte fastställts.Läkarna diskuterar fortfarande genomförbarheten av livslång dietterapi, men de håller med om att en diet på minst 18 bör följas.
Fenylketonuri som diagnostiserats hos en kvinna är inte en anledning att vägra födelse av ett barn.Förväntade mödrar med PKU för att förhindra fosterskada under graviditeten och för att förhindra möjliga komplikationer är det nödvändigt att observera en diet med en fenylalanin-begränsad diet (vid leveransen) (upp till 242 μmol /l).
Laktosfria blandningar för spädbarn
Kostholdet för fenylketonuri är baserat på en betydande minskning av mängden naturligt protein i den dagliga kosten, men barnets kropp kan inte utvecklas normalt i frånvaro av nödvändiga spårelement.För att tillgodose barnets behov av protein används laktosfria aminosyrablandningar, som enligt rysk lag måste tillhandahållas gratis.
Spädbarns tolerans för fenylalanin förändras snabbt under det första leveåret, så dess koncentration i barnets blod måste övervakas och kosten justeras.Blandningarna är utformade för vissa åldersgrupper:
- spädbarn under ett år föreskrivs Afenilak 15, Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- Barn äldre än 1år, utnämna berikade med vitaminer och mineralblandningar med högt proteininnehåll - PKU Prima, P-AM Universal, PKU-1, PKU-2, Maximeid XP, Maximum XP.

Proteintillskott livsmedel
En av de viktigaste komponenterna i en diet för fenylketonuri är lågproteinstärkelsebaserade produkter.Dessa tillskott innehåller kaseinhydrolysat, tryptofan, tyrosin, metionin, kväve och ger barnets dagliga behov av det protein som behövs för normal utveckling och tillväxt.Specialiserade livsmedel som fyller bristen på essentiella mineraler och aminosyror när de saknar kost är:
- Berlofen;
- Zimorgan;
- Minafen;
- Aponti.
Kost för förskolebarn och skolbarn
När kroppen anpassar sig till fenylalanin kan barn från 5 års ålder gradvis minska sina kostbegränsningar.En utvidgning av en diet sker genom introduktion av spannmål, mejeriprodukter, köttprodukter.Äldre studenter har redan en hög tolerans mot fenylalanin, så i denna ålder kan du fortsätta att utöka kosten, samtidigt som du övervakar svaret på eventuella näringsförändringar.Följande metoder används för att kontrollera barnets tillstånd:
- bedömning av neurologiska parametrar, psykologiskt tillstånd;
- kontroll av prestanda för elektroencefalogram;
- bestämning av fenylalaninnivå.
Grupper av livsmedel med PKU
Diet för patienter med PKU, tillsammans med lågt proteinstärkelseprodukter och läkemedelsblandningar inkluderar även produkter av naturligt ursprung.Vid upprättandet av menyn bör mängden protein som konsumeras tydligt beräknas och den dos som rekommenderas av läkaren bör inte överskridas.För att eliminera toxiska effekter på kroppen har 3 listor med produkter utvecklats som innehåller förbjudna (röda), inte rekommenderade (orange) och tillåtna (gröna) föremål.
Rödlista
Fenylketonuria utvecklas på bakgrund av frånvaron av ett enzym som konverteras till tyrosinfenylalanin, så ett högt proteininnehåll är grunden för att lista produkterna i den förbjudna (röda) listan.Artiklarna i denna lista bör helt utesluta dieten hos en patient med PKU:
- kött;
- inre organ hos djur, biprodukter;
- korv, korv;
- skaldjur (inklusive fisk);
- ägg från alla fåglar;
- mejeriprodukter;
- nötter;
- baljväxter och spannmål;
- sojaprodukter;
- gelatininnehållande rätter;
- konfekt;
- aspartam.
Orange lista
Produkter som ska doseras till ett barn som diagnostiserats med PKU ingår i den orange listan.Inkluderingen i kosten för artiklarna i denna lista är acceptabel, men i ett strikt begränsat antal.Även om dessa produkter inte innehåller mycket protein, kan de också öka nivån av fenylalanin, så deras användning rekommenderas inte:
- konserverade grönsaker;
- potatis- och risrätter;
- kål;
- mjölk;
- sherbet.
Grön lista
Proteinfria produkter får användas till patienter med en diagnos av fenylketonuri utan begränsning.Innan du köper föremålen på den gröna listan är det nödvändigt att undersöka kompositionen som anges på förpackningen och se till att det inte finns något färgämne för aspartam som innehåller fenylalanin:
- frukt;
- grönsaker (utom potatis och kålar);
- bär;
- greener;
- stärkelsefull spannmål (sago);
- honung, socker, sylt;
- mjölprodukter tillverkade av majs eller rismjöl;
- oljor, fetter (smör, solros, oliv).
Hur man styr nivån av fenylalanin i blodet
Fenylketonuria är en obotlig sjukdom som kan överföras till stagnationsfasen genom användning av diet och terapeutiska åtgärder.Vid förändring av levnadsförhållanden, störning av en kost, kan sjukdomen förvärras igen, därför behöver patienter livslång observation.Kontrollprocessen är att regelbundet bestämma nivån av fenylalanin i blodet.Analysfrekvensen beror på patientens ålder:
- upp till 3 månader - blodundersökning bör göras varje vecka för att få konsekventa resultat;
- från 3 månader till 1 år - 1-2 gånger i månaden;
- 1 till 3 år - en gång varannan månad;
- äldre än 3 år - kvartalsvis.

Blod för analys ges 3-4 timmar efter intag.Förutom screening styrs utvecklingen av PKU genom att bestämma näringsstatus, fysisk, emotionell utveckling hos patienten, nivån på intellektuella förmågor.och språkutveckling.Observationer kan kräva ytterligare diagnostik med deltagande av lämpliga specialister.
Video
Informationen som presenteras i denna artikel är endast för vägledning.Artikeln kräver inte självbehandling.Endast en kvalificerad läkare kan diagnostisera och rekommendera behandling baserat på den enskilda patientens individuella egenskaper.















